
Veröffentlicht am20 May 2024
min lesen
Ansichten
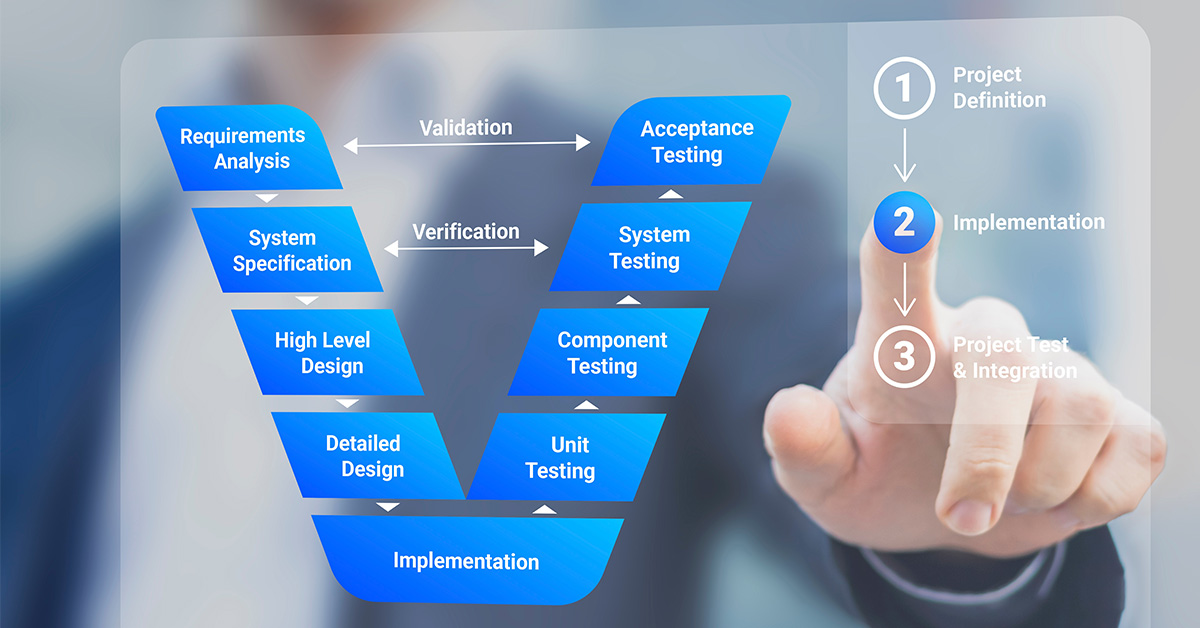
Die Herstellung von Medizinprodukten ist durch ein strenges regulatorisches Umfeld gekennzeichnet, das ein Höchstmaß an Produktwirksamkeit und Patientensicherheit gewährleisten soll. Von zentraler Bedeutung für die erfolgreiche Bewältigung dieses Umfelds sind Designverifizierung und -validierung (V&V), die sowohl bei der Produktentwicklung als auch bei der Einhaltung von Vorschriften eine entscheidende Rolle spielen.
Design V&V sind systematische Ansätze, die in der Medizinprodukteindustrie angewandt werden, um sicherzustellen, dass ein Produkt die Anforderungen des Endverbrauchers erfüllt und alle gesetzlichen Vorschriften einhält. Die Verifizierung bestätigt, dass die Ergebnisse der Entwicklung den Anforderungen entsprechen, während die Validierung sicherstellt, dass das Produkt den festgelegten Verwendungszwecken entspricht. Beide Prozesse sind integraler Bestandteil des Produktlebenszyklus - sie fördern die Innovation, gewährleisten die Sicherheit und halten die Vorschriften ein.
Der weltweite Markt für Medizinprodukte wird von verschiedenen Aufsichtsbehörden geregelt, von denen jede ihre eigenen Anforderungen an die Gewährleistung der Produktsicherheit und -wirksamkeit stellt. Die U.S. Food and Drug Administration (FDA) und die Medizinprodukteverordnung der Europäischen Union (EU MDR) sind zwei dieser Einrichtungen, die strenge Standards für Medizinprodukte festlegen. Die Einhaltung dieser Standards durch eine wirksame V&V erleichtert nicht nur den Marktzugang, sondern verringert auch das Risiko von Problemen nach der Markteinführung erheblich, was die Sicherheit der Patienten und die Zuverlässigkeit der Produkte erhöht.
Verstehen der regulatorischen Anforderungen (FDA, EU MDR, etc.)
Um die Einhaltung international anerkannter Normen wie FDA 21 CFR 820.30 (Design Control) und ISO 13485 7.3 (Design und Entwicklung) zu gewährleisten, ist es für Hersteller unerlässlich, die unterschiedlichen, sich jedoch überschneidenden Anforderungen dieser Regelwerke genau zu verstehen. Beide Regelwerke legen großen Wert auf die Notwendigkeit einer umfassenden Dokumentation, die während des gesamten Konstruktions- und Entwicklungsprozesses sorgfältig geführt werden muss. Die Hauptunterschiede zwischen den beiden Regelwerken ergeben sich jedoch aus ihrer geografischen Anwendbarkeit und den Besonderheiten ihrer Ausführung, die je nach dem regulatorischen Umfeld variieren können. Das Verständnis dieser Nuancen ist für Hersteller von entscheidender Bedeutung, um sich in der komplexen Landschaft der globalen Konformität zurechtzufinden und so sicherzustellen, dass ihre Produkte die höchsten Sicherheits- und Wirksamkeitsstandards erfüllen.
Risikomanagement bei der Verifizierung und Validierung
Das Risikomanagement spielt in den V&V-Prozessen eine entscheidende Rolle, denn es verlangt von den Herstellern, dass sie potenzielle Risiken im Zusammenhang mit ihren Medizinprodukten proaktiv erkennen und mindern. Durch die Anwendung eines risikobasierten Ansatzes von den ersten Entwicklungsphasen an können die Hersteller nicht nur die Einhaltung strenger gesetzlicher Normen gewährleisten, sondern auch der Patientensicherheit in jeder Phase des Produktlebenszyklus Vorrang einräumen. Dies beinhaltet eine gründliche Bewertung potenzieller Gefahren und ihrer Auswirkungen und stellt sicher, dass jeder Aspekt des Produktdesigns und der Funktionalität auf Sicherheit geprüft wird, bevor es auf den Markt kommt.
Integration von Human Factors Engineering
Durch die Integration von Human Factors Engineering (HFE)-Prinzipien in die V&V-Prozesse können die Entwickler sicherstellen, dass die Medizinprodukte bei der Bedienung durch den Endanwender wie vorgesehen funktionieren. Diese Integration erhöht nicht nur die Gerätesicherheit, indem sie die Wahrscheinlichkeit von Benutzerfehlern verringert, sondern verbessert auch die allgemeine Benutzerfreundlichkeit, indem sie die Geräte intuitiver und benutzerfreundlicher macht. Folglich trägt die Anwendung der HFE-Prinzipien in der Entwicklungsphase wesentlich zum erfolgreichen Einsatz von Medizinprodukten bei, was letztlich zu besseren Ergebnissen und höherer Zufriedenheit der Patienten führt.
Die Landschaft der Medizinprodukteherstellung entwickelt sich ständig weiter, angetrieben durch technologische Fortschritte und sich ändernde gesetzliche Anforderungen.
Eine wirksame Designverifizierung und -validierung ist für den Erfolg von Medizinprodukteherstellern auf dem globalen Markt von grundlegender Bedeutung. Durch die Einhaltung gesetzlicher Vorschriften, die Anwendung von Risikomanagementstrategien und die Integration von Human Factors Engineering können Unternehmen sicherstellen, dass ihre Produkte effektiv, sicher und konform sind. Angesichts der Tatsache, dass in der Medizinprodukteindustrie weitere Innovationen und regulatorische Entwicklungen anstehen, wird es für Hersteller weltweit weiterhin von entscheidender Bedeutung sein, mit den besten Praktiken im Bereich V&V Schritt zu halten.
Sind Sie bereit, sicherzustellen, dass Ihr Medizinproduktdesign den wichtigsten Vorschriften entspricht? Starten Sie noch heute Ihren Weg zur Konformität. Erschließen Sie den Erfolg Ihres Produkts mit unserer Hilfe - beginnen Sie jetzt damit.



