
公開日20 May 2024
最小読み取り時間
ビュー
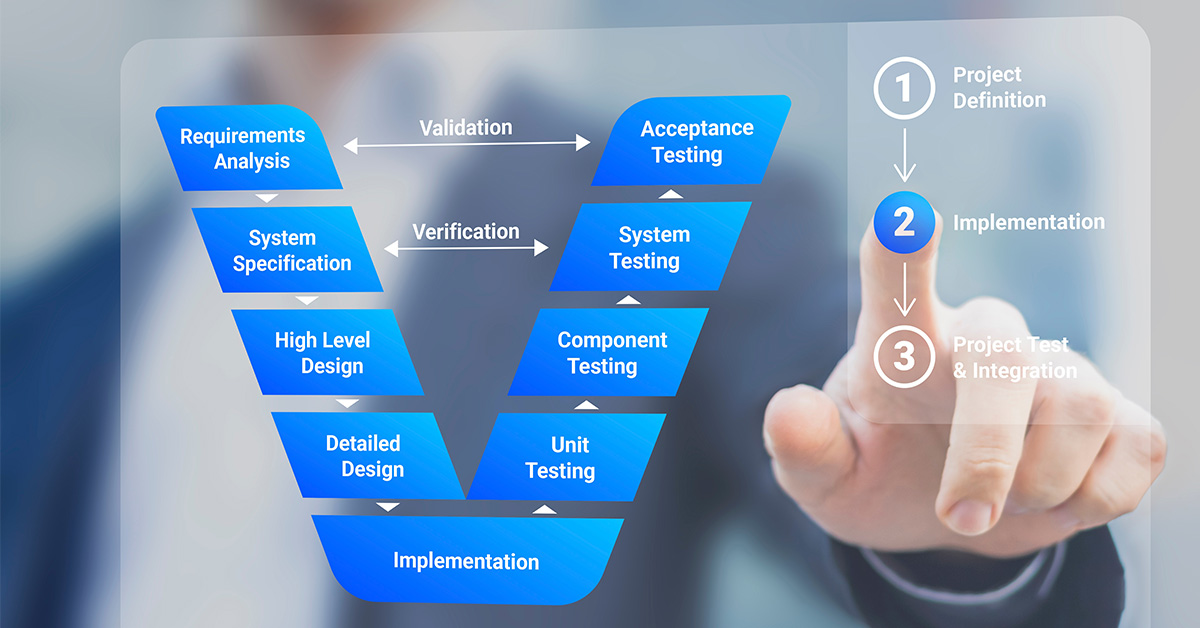
医療機器製造の現場は、最高レベルの製品の有効性と患者の安全性を確保するために設計された厳格な規制環境によって特徴付けられる。このような環境をうまく乗り切るための中心的な役割を果たすのが、製品開発と規制遵守の両面で重要な柱となる設計検証・妥当性確認(V&V)です。
設計V&Vは、製品がエンドユーザーのニーズを満たし、すべての規制要件に準拠していることを確認するために、医療機器業界で利用されている体系的なアプローチです。検証は、設計出力が入力要件を満たしていることを確認し、検証は、機器が定義された使用目的に適合していることを確認する。両プロセスは、イノベーションを促進し、安全性を確保し、コンプライアンスを維持するという製品ライフサイクルに不可欠なものである。
世界の医療機器市場は様々な規制機関によって管理されており、それぞれが製品の安全性と有効性を確保するための要求を持っている。米国食品医薬品局(FDA)と欧州連合(EU)の医療機器規制(EU MDR)は、医療機器の厳格な基準を定めている2つの団体です。効果的なV&Vを通じてこれらの基準を遵守することは、市場参入を容易にするだけでなく、市販後の問題のリスクを大幅に低減し、患者の安全性と製品の信頼性を強化します。
規制要件(FDA、EU MDRなど)の理解
設計管理に焦点を当てたFDA 21 CFR 820.30や、設計開発に関するISO 13485の条項7.3など、国際的に認知された規格に確実に準拠するためには、メーカーはこれらの規制の枠組みによって概説された、異なるが重複する要件を完全に理解することが不可欠である。両規制は、設計・開発プロセス全体を通じて細心の注意を払い維持されなければならない包括的な文書化の必要性を強く強調している。しかし、この2つの主な違いは、地理的な適用可能性と、規制環境によって異なる可能性のある具体的な実施内容に起因している。メーカーがグローバルコンプライアンスの複雑な状況を乗り切るには、このようなニュアンスを理解することが極めて重要であり、その結果、製品が最高水準の安全性と有効性を満たすことが保証される。
検証・バリデーションにおけるリスク管理
リスク管理は、V&Vプロセスにおいて重要な役割を果たし、製造業者は医療機器に関連する潜在的なリスクを積極的に特定し、軽減することが義務付けられています。開発の初期段階からリスクベースのアプローチを採用することで、メーカーは厳格な規制基準への準拠を保証できるだけでなく、製品のライフサイクルの各段階において患者の安全を優先することができる。これには、潜在的な危険性とその影響の徹底的な評価が含まれ、製品の設計と機能のあらゆる側面が、市場に出る前に安全性について精査されることが保証される。
ヒューマン・ファクター・エンジニアリングの統合
ヒューマンファクター工学(HFE)の原則をV&Vプロセスに組み込むことにより、開発者は、エンドユーザーが操作する際に医療機器が意図したとおりに動作することを保証することができます。この統合は、ユーザーエラーの可能性を低減することで機器の安全性を高めるだけでなく、全体的な使いやすさを向上させ、機器をより直感的でユーザーフレンドリーなものにします。その結果、開発段階におけるHFEの原則の適用は、医療機器の展開の成功に大きく貢献し、最終的に患者の転帰と満足度の向上につながります。
医療機器製造の状況は、技術の進歩と変化する規制要件によって絶えず進化しています。
効果的な設計検証と妥当性確認は、グローバル市場で医療機器メーカーが成功するための基盤です。規制要件を遵守し、リスク管理戦略を採用し、ヒューマンファクター工学を統合することで、企業は製品の有効性、安全性、コンプライアンスを確保することができます。医療機器業界がさらなる革新と規制の進化を遂げる中、V&Vのベストプラクティスに遅れを取らないことは、世界中のメーカーにとって引き続き重要です。
医療機器の設計が主要な規制に準拠していることを確認する準備はできていますか?今すぐコンプライアンスへの道を歩み始めましょう。当社の支援で製品の成功を引き出しましょう。



